

La Sté Aid’ARC est implantée sur 2 régions :
- en Normandie (proche de PARIS)
- et dans le Sud-Ouest (proche de TOULOUSE)
Nous intervenons sur toute la France et principalement sur les villes suivantes :
La Sté Aid’ARC est implantée sur 2 régions :
Nous intervenons sur toute la France et principalement sur les villes suivantes :
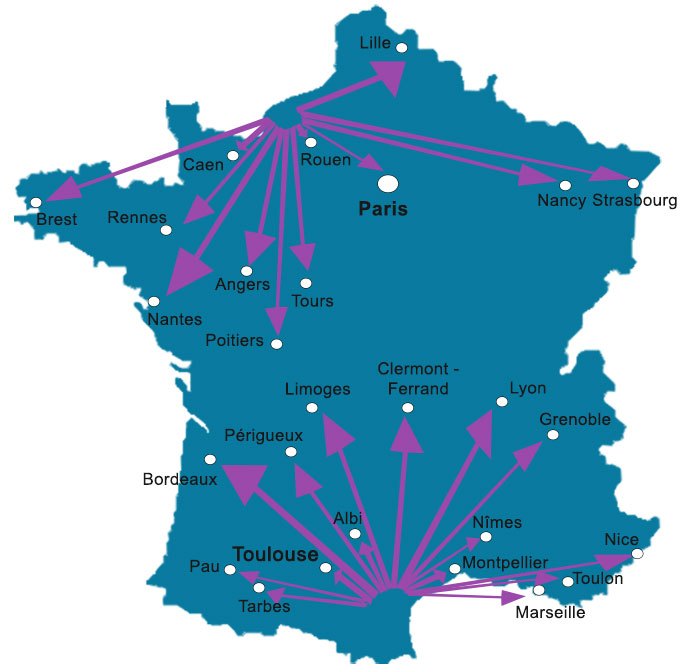
Déplacements sur toute la France métropolitaine et les DOM-TOM.
Nous intervenons aussi en Europe et à l’international dans des pays comme la Belgique, l’Angleterre, la Suisse, l’Espagne, l’Allemagne et l’Australie.